ทวีชัย วิทิตสุวรรณกุล
อาจารย์ ดร.
ข้อมูลติดต่อ
- ภาควิชาเคมี
- ห้อง : C607A
- โทรศัพท์ : 02-201-5170
- E-Mail: taveechai.wit@mahidol.ac.th
การศึกษา
- Ph.D., Texas A&M University, USA
- Postdoctoral Research Fellow, University of Michigan, USA
ความเชี่ยวชาญ
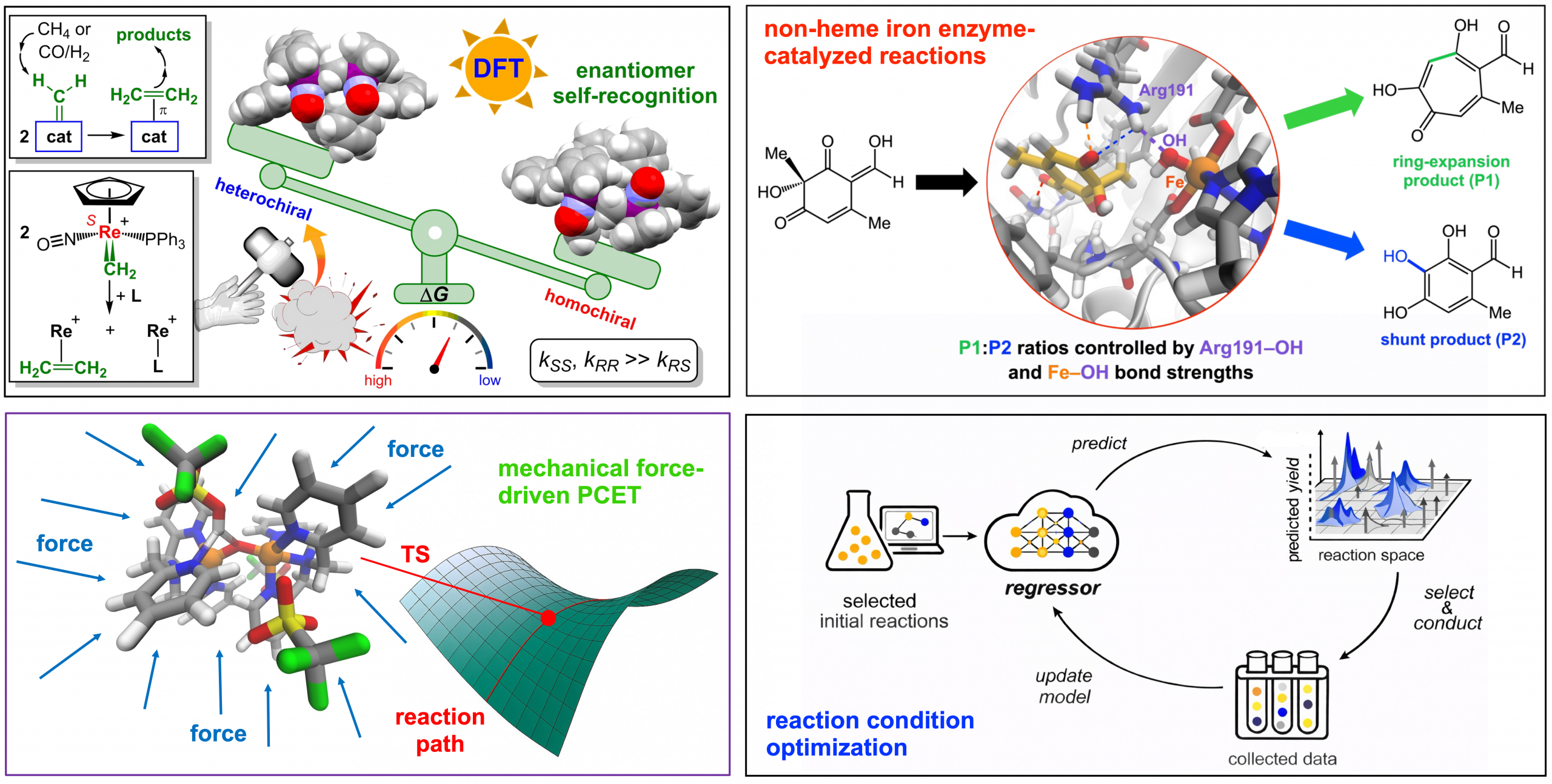
- Quantum Chemical Simulations and Multiscale Modeling: Applying state-of-the-art quantum chemical simulations and multiscale computational approaches to elucidate reaction mechanisms and origins of selectivity in complex chemical and biological systems, spanning small molecules (such as homogeneous catalysts and transition-metal complexes) to macromolecular systems (such as metalloenzymes and functional polymers)
- Mechanistic Modeling of External Stimuli-Driven Reactions: Developing computational frameworks to elucidate reactions under external stimuli, including mechanical force, enabling predictive understanding of unconventional reaction pathways
- Non-Covalent Interactions and Enantioselectivity: Elucidating the electronic and stereochemical origins of enantioselective reactions and enantiomer self-recognition driven by non-covalent interactions, guiding rational catalyst and molecular design.
- Machine Learning-Driven Reaction Discovery: Integrating machine learning with reaction path simulations and reaction condition optimization to accelerate catalyst design, improve efficiency, and reduce experimental resource consumption
Research Interest
Our computational chemistry research group focuses on simulating reactions for understanding complex chemical and biological phenomena. We implement quantum chemical simulations with multiscale modeling and machine learning to simulate and investigate complex challenging systems in order to drive advances in catalyst design, reaction discovery, and sustainable synthesis processes.
Our research spans diverse applications, including mechanistic studies of bimolecular coupling of methylidene to ethylene ligands in both coordinatively saturated and unsaturated transition-metal methylidene complexes; investigations of non-heme iron enzyme-catalyzed reactions involved in natural product biosynthesis; mechanical force-induced proton-coupled electron transfer in biomimetic transition-metal complexes; and machine learning-driven reaction path simulations and reaction condition optimization. Across these areas, we aim to apply and develop predictive computational tools to accelerate catalyst development and guide experimental design.