
Phiphob Naweephattana,† Boodsarin Sawatlon,† and Panida Surawatanawong*,†,‡
†Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Mahidol
University, Bangkok 10400, Thailand
‡Center of Sustainable Energy and Green Materials, Mahidol University, Salaya, Nakhon Pathom 73170, Thailand
*E-mail: panida.sur@mahidol.ac.th
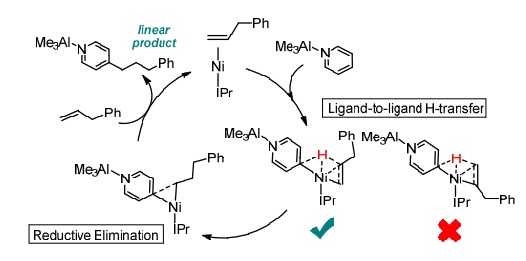
The hydroheteroarylation of allylbenzene with pyridine as catalyzed by Ni/AlMe3 and Nheterocyclic carbene ligand has recently been established. Density functional calculations revealed that the common stepwise pathway, which involves the C-H oxidative addition of pyridine-AlMe3 before the migratory insertion of allylbenzene, is unlikely as the migratory insertion needs to overcome a prohibitively high energy barrier. In contrast, the ligand-to-ligand hydrogen transfer (LLHT) pathway is more favorable, in which the hydrogen is transferred directly from the para-position of pyridine-AlMe3 to C2 of allylbenzene. Our distortion-interaction analysis and natural bond orbital analysis indicate that the interaction energy is strongly correlated with the extent of the charge transfer from the alkene (hydrogen acceptor) to the pyridine-AlMe3 (hydrogen donor), which dictates the selectivity of the H-transfer to the C2 position of allylbenzene. Then, the subsequent C-C reductive elimination of the regioselective linear product is facilitated by the steric hindrance of the IPr ligand. Understanding these key factors affecting the product regioselectivity is important to the development of catalysts for hydroheteroarylation of alkenes.
11340-11349. https://pubs.acs.org/doi/10.1021/acs.joc.0c01449